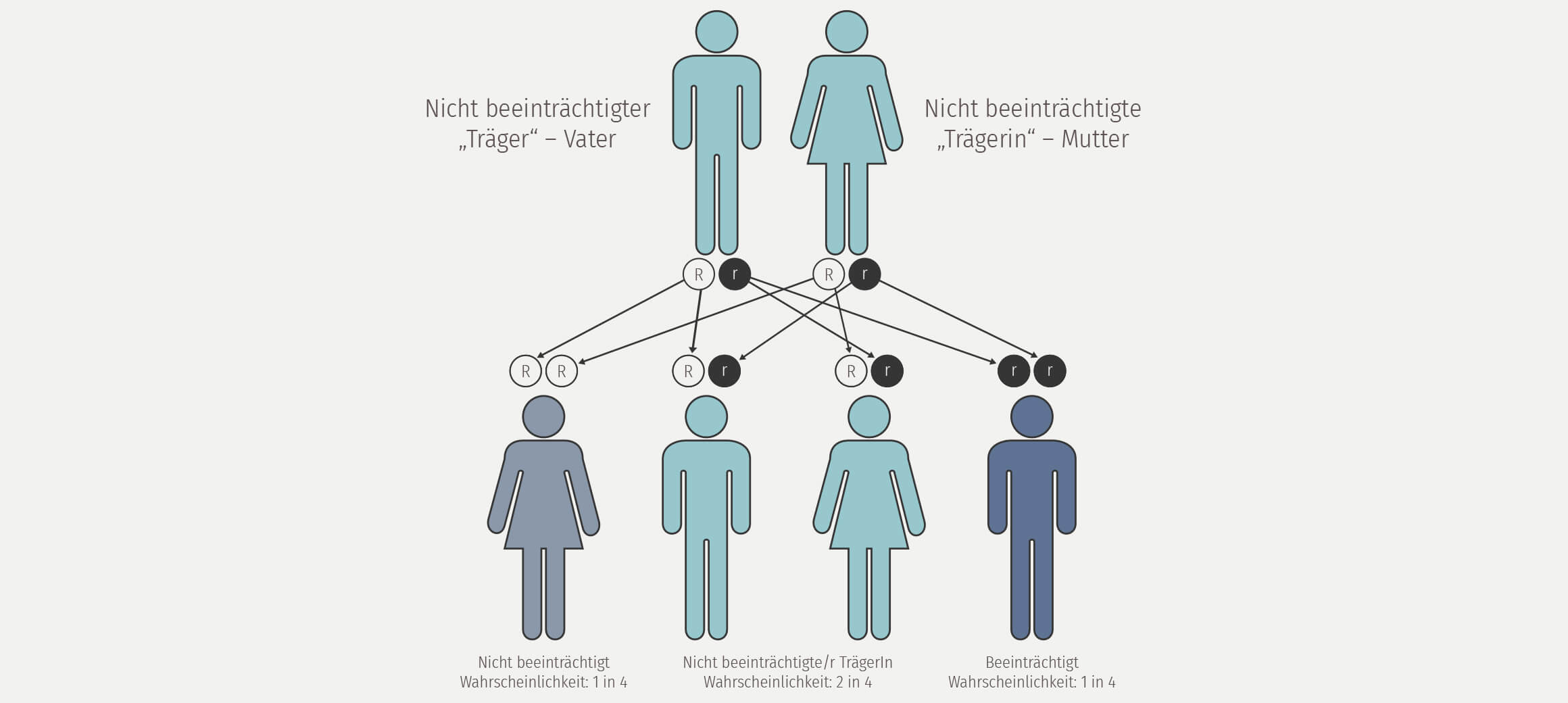
Das Usher-Syndrom ist ein sowohl klinisch wie genetisch uneinheitliches Krankheitsbild, das erstmals 1858 von dem deutschen Augenarzt von Graefe beschrieben wurde als Kombination von Innenohrschwerhörigkeit und der Augenerkrankung Retinitis pigmentosa (Link zu Retinitis pigmentosa ). Charles Usher zeigte 1914 als erster die Erblichkeit der Erkrankung auf. Doch erst Hallgren untersuchte systematisch die Häufigkeit des Usher Syndroms in Schweden und erkannte die Einteilung in 2 Usher Typen (schwerhörig und taub). Heute sind 3 verschiedene Subtypen des Usher Syndroms bekannt.
Usher Typ I (USH1)
Der Typ I des Usher-Syndroms stellt die schwerere Verlaufsform dar. Betroffene sind von Geburt an gehörlos. Ausserdem bestehen starke Gleichgewichtsstörungen, die Gleichgewichtsorgane funktionieren nicht. Die Augenerkrankung beginnt in der frühen Kindheit (1. Lebensjahrzehnt) mit Nachtblindheit und Gesichtsfeldeinschränkungen.
Usher-Syndrom Typ II (USH2)
Die Krankheit verläuft sowohl was die Seh- als auch die Hörbehinderung betrifft deutlich milder als beim Typ I. Es liegt eine mittel- bis hochgradige Schwerhörigkeit ab der Geburt vor. Zu einem Fortschreiten der Schwerhörigkeit kommt es nicht. Der Gleichgewichtssinn ist normal. Die Augenerkrankung bemerkt der Betroffene häufig erst im frühen Erwachsenenalter.
Usher-Syndrom Typ III (USH3)
Menschen mit Usher Syndrom Typ III werden schwerhörig geboren. Erst im fortgeschrittenen Alter werden sie taub. Exakte Aussagen zum Gleichgewichtsinn liegen bislang nicht vor. Die RP verläuft wie beim Typ II. Insgesamt ist der Typ III sehr selten und nur in Finnland und in wenigen nach den USA ausgewanderten finnischen Familien beschrieben worden.
Wichtig zu beachten ist, dass die Hör- und Seherkrankung beim Usher-Syndrom zu unterschiedlichen Zeitpunkten auftritt. Die Hörerkrankung beginnt beim Usher-Syndrom Typ I und II nach der Geburt. Beim Typ III ist das Hörvermögen anfangs noch erhalten, im zweiten Lebensjahrzehnt werden die Betroffenen taub. Die Seherkrankung beginnt beim USH1 in den ersten 10 Lebensjahren und beim USH2 und USH3 meist bis zum Ende des 20. Lebensjahres.